一种更加准确和快速的对接方法:教程:用AutoDock Vina进行分子对接
本文讲述大分子受体与小分子配体的分子对接过程。只是记录一遍流程,该扩展的地方如果有空再写文详述。
1.软件安装及工作目录创建
下载安装包MGLTools1.5.6(当前版本,2017年12月16日),按照提示安装即可。注意安装的时候地址中不要含有中文,以免不可知的问题。安装好之后,在安装目录下找到adt.bat,右键发送一个快捷方式到桌面。

比如安装目录为:D:Program Files (x86)The Scripps Research Institute ,那么adt.bat文件则位于D:Program Files(x86)The Scripps Research InstituteMGL下。同时为了后续方便,我们在E盘下创建一个工作目录E:AutoDock。创建好之后,把MGLtools安装目录下的Autodock4.2.6文件夹复制到工作目录中(最终如下图)。

回到刚才创建的桌面快捷方式,右键,设置起始位置为E盘的工作目录E:AutoDock

好了,工作目录就设置完毕了。
MGLTools 1.5.6安装包下载地址:http://mgltools.scripps.edu/downloads
另外还需要Pymol软件(查看3D结构),Open Babel(用来将pdbqt文件转成pdb文件,便于查看),下载地址自行查找。
2.受体和配体的pdb文件准备
以PDB数据库的3uf0(这是PDBID)为例,这是某个脱氢酶与NADP的复合物结构。我事先用Pymol软件将受体和配体分子的结构从3uf0中提取出来,分别保存为r.pdb和nap.pdb。水分子已经在pymol软件中全都删除了。“受体”和“配体”的叫法不准确,但是为了方便,我就先这么称呼了。
如果是自己实验室解析出来的结构,那么需要手动在pymol中去掉水分子;另外,一个晶格中可能会有好几个分子,表现为“N聚体”。此时也要把其他的分子都删掉,只保留一个拷贝。比如3uf0有A/B两条链,我们只要其中一个就可以了。除非这个结合位点是有AB两条链共同组成。为了方便大家,我把这几个文件都放出来。3uf0.pdb, r.pdb, nap.pdb
3. 受体和配体的pdbqt文件准备
我们把r.pdb和nap.pdb拷贝到工作目录中。双击创建的快捷方式“adt.bat- 快捷方式”,
3.1受体的准备
打开AutoDockTools,菜单栏“File->Read Molecule”打开r.pdb
3.1.1除水,之前在pymol中已经把水分子全都删了,这一步省略
3.1.2加氢。菜单栏“Edit->Hydrogens->Add”直接点击OK就行。
3.1.3计算电荷。菜单栏“Edit->Charges->Compute Gasteiger”
因为我们这里做的是大分子受体与小分子配体之间的对接,一般认为大分子为刚性的,而小分子是柔性的。所以还需要对受体分子做个操作,即:
3.1.4添加原子类型。菜单栏“Edit->Atoms->Assign AD4 type”
3.1.5保存。菜单栏“File->Save->Write PDBQT”,此时可在工作目录下看到多了一个“r.pdbqt”文件
3.2配体的准备
好了,我们可以在Dashboard中把受体的分子给删了(右键->delete)。
通过ADT菜单栏“Ligand->Input->Open..”打开nap.pdb文件,弹出一个对话框,点击‘确定’

注意,这个对话框里面有个提示,就是配体分子的总电荷数不是整数。所以要调整下,菜单栏:“Edit->Charges->Check Totals on Residues”,然后点击“Spread Charge Deficit over all atoms in residue”,然后“Dismiss”
3.2.1判定配体的root。ADT菜单栏:Ligand->Torsion Tree->Detect Root
3.2.2选择配体可扭转的键。Ligand->Torsion Tree->Choose Torsions
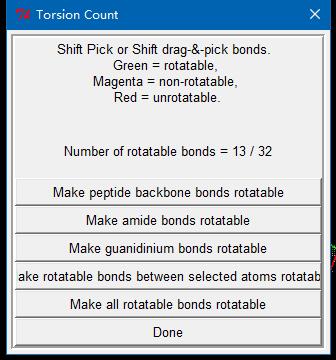
表示该分子32个键中有13个可旋转的。其中红色表示不可旋转的键,绿色表示可旋转键,紫色表示不可扭转,通常为肽键,点击“Done”即可。如果要设置某个键不可扭转,那么先按住shift键,然后鼠标单击即可(颜色变成紫色)。
3.2.3保存。ADT菜单栏:“Ligand->Output->Save as PDBQT”
4.Grid
DashBoard中删掉配体分子。
4.1 打开受体分子,ADT菜单栏:“Grid->Macromocule->Open”,打开“r.pdbqt”。弹出的对话框,点击Yes及确定
4.2 打开配体分子,ADT菜单栏:“Grid->Set Map Types->Open ligand”
4.3 设置GridBox。ADT菜单栏:“Grid->Grid Box…”,打开Grid Options对话框。调整X,Y,Z及格子中心坐标。我们这里就设置成如下图:
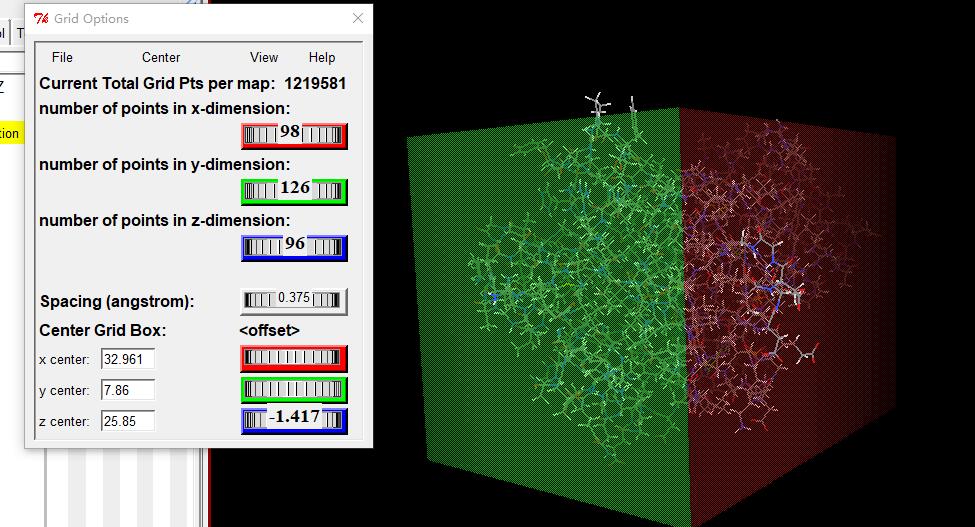
设置完成后点击Grid Options 菜单中:File→Close saving current 保存并关闭对话框。
4.4保存gpf文件。ADT菜单栏:“Grid->Output->Save GPF”,对话框中,就写g.gpf做为文件名。注意:不要省略后缀名。
5. 运行autogrid
ADT菜单栏:Run->Run Autogrid
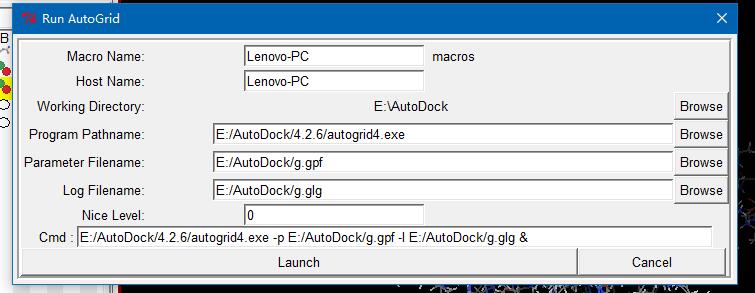
有个小弹窗出来,等待它消失就好了,此时工作目录会多出一堆文件。
6. 准备DPF文件
在Dashboard中删掉受体和配体分子。
6.1打开受体分子。ADT菜单栏:“Docking->Macromocule->Set rigid macromocule”,选择r.pdbqt
6.2打开配体分子。ADT菜单栏:“Docking->Ligand->Open”,选择nap.pdbqt。对话框选择Accept
6.3设置算法。ADT菜单栏:“Docking->Search Parameters->Genetic Algorithm”,就默认设置好了,点击Accept
6.4 设置对接参数。ADT菜单栏:“Docking->Docking Parameters”,就默认设置好了,点击Accept
6.5 保存。ADT菜单栏:“Docking->Output->Genetic Algorithm”,文件名就写d.dpf。注意:加上后缀名
7. 对接
ADT菜单栏:“Run->Run AutoDock”如下图所示,点击Launch。
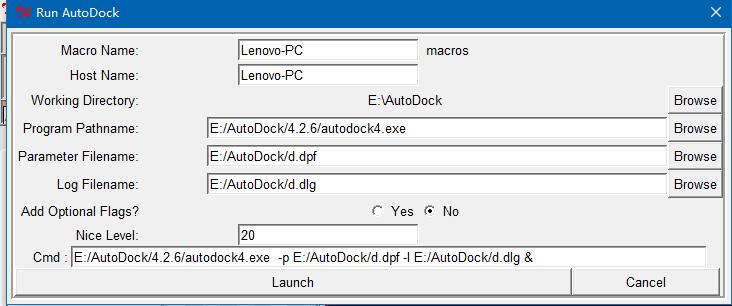
又弹出一个小框框,等他消失。发现多了一个dlg文件,这就是对接的最终结果,一般有10种对接方式。
8.查看对接结果。ADT菜单栏:“Analyze->Open”打开刚才生成的.dlg文件。加上受体大分子,ADT菜单栏:“Analyze->Macromolecule->Open”。一个个的查看刚才对接的结果,ADT菜单栏:“Analyze->Conformations->Play”。
9.导出对接结果并转换成PDB格式的数据。我们以第一个结果为例,点击像“&”符号的按钮(如下图),然后再点击“Write Complex”按钮生成pdbqt文件,我们就命名为”c.pdbqt”。注意不要忘了后缀名。打开Open Babel软件,找到刚才的c.pdbqt,以及写好输出的pdb文件名,然后点击CONVERT按钮。


附:显示配体结合口袋的pymol命令
create pocket, byres 3uf0 within 5 of resn nap
#这条命令的目的就是以配体nap为中心,在3uf0这个蛋白质里找出跟nap距离在5 A的氨基酸,生成一个pocket。
show sticks, pocket
#将pocket的残基显示成棒状。
hide lines
#隐藏蛋白质的line,这样pymol只显示pocket的残基
Souce: 纽普生物 2017-12-27