师妹:师兄,我想建个树。
师兄:啥树啊!你家的family tree啊?
师妹:师兄,你不要调戏我,我就建一个简单的进化树!这个怎么做啊?
简单呀,你可以用MEGA,先去MEGA官网(http://www.megasoftware.net/)下载这个软件,免费的啊。MEGA(Molecular Evolutionary Genetics Analysis )是一个功能非常强大的分子进化遗传分析软件,可用于序列比对、进化树的推断、估计分子进化速度、验证进化假说等。下面师兄给你详细介绍如何利用Mega软件构建进化树。
1.首先将需要进行建树的序列保存为fasta格式,并将文件扩展名改为.fasta。
.fasta序列格式以“>”开头。“>”后面这一行写名称,回车,下一行写序列,氨基酸序列类似,所有序列保存在一个txt文件中。例如:
>gene1/speciesname NCBI accession number
ATCGGCGTAGCTAGATGCTAGTATCGTA
>gene1/speciesname NCBI accession number
AGTAGCTAGTGATGTA
2. 点击Align--Edit/built Alignment,选择创建一个新的比对,点OK
根据要求选择DNA或者蛋白质序列
3.打开需要比对的.fasta文件
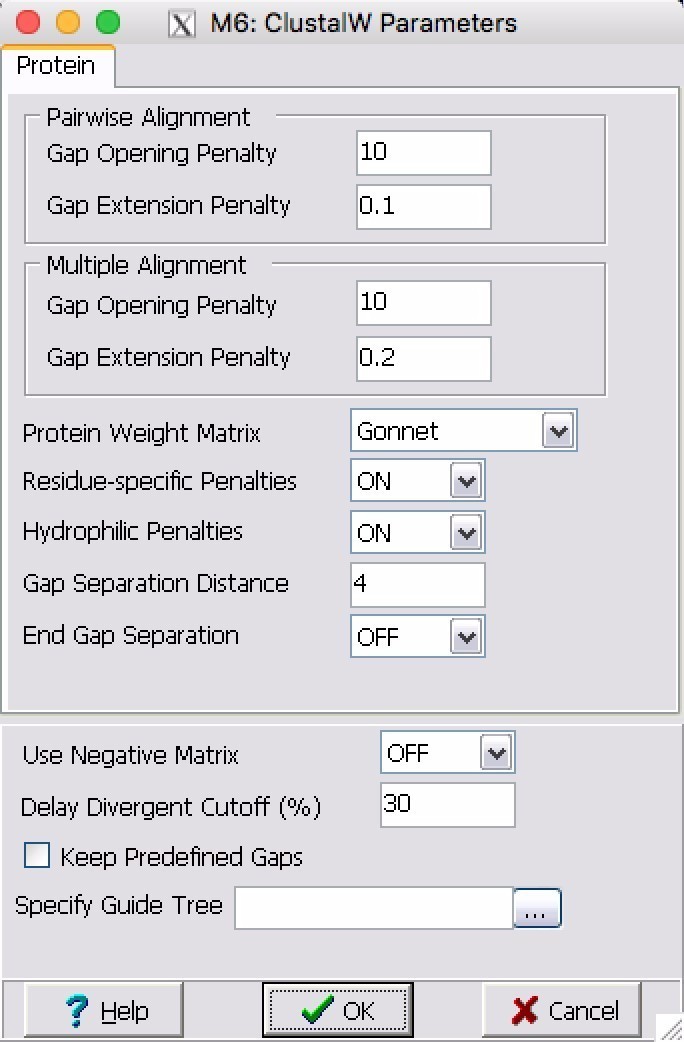
4. 点击Alignment-Align by Clustal W,选择所有序列,出现下图,所有参数为默认,点击OK。
5. 我们看到在未对齐之前,由于序列长度不一样,有些序列长出来很多,而有的序列在这些位点全是gap,为了排除gap位点的干扰。我们需要将序列两端对齐。两端以比对上最短的序列为准,删除其他序列5’和3’多余的部分,可以看到在序列比对上的部分,最上面一行软件标记为“*”,我们需要将没有标记“*”的位点删除,可以用shift一起选择没有标记“*”开始和末端的位点,选好后点击鼠标右键,单击delete删除。
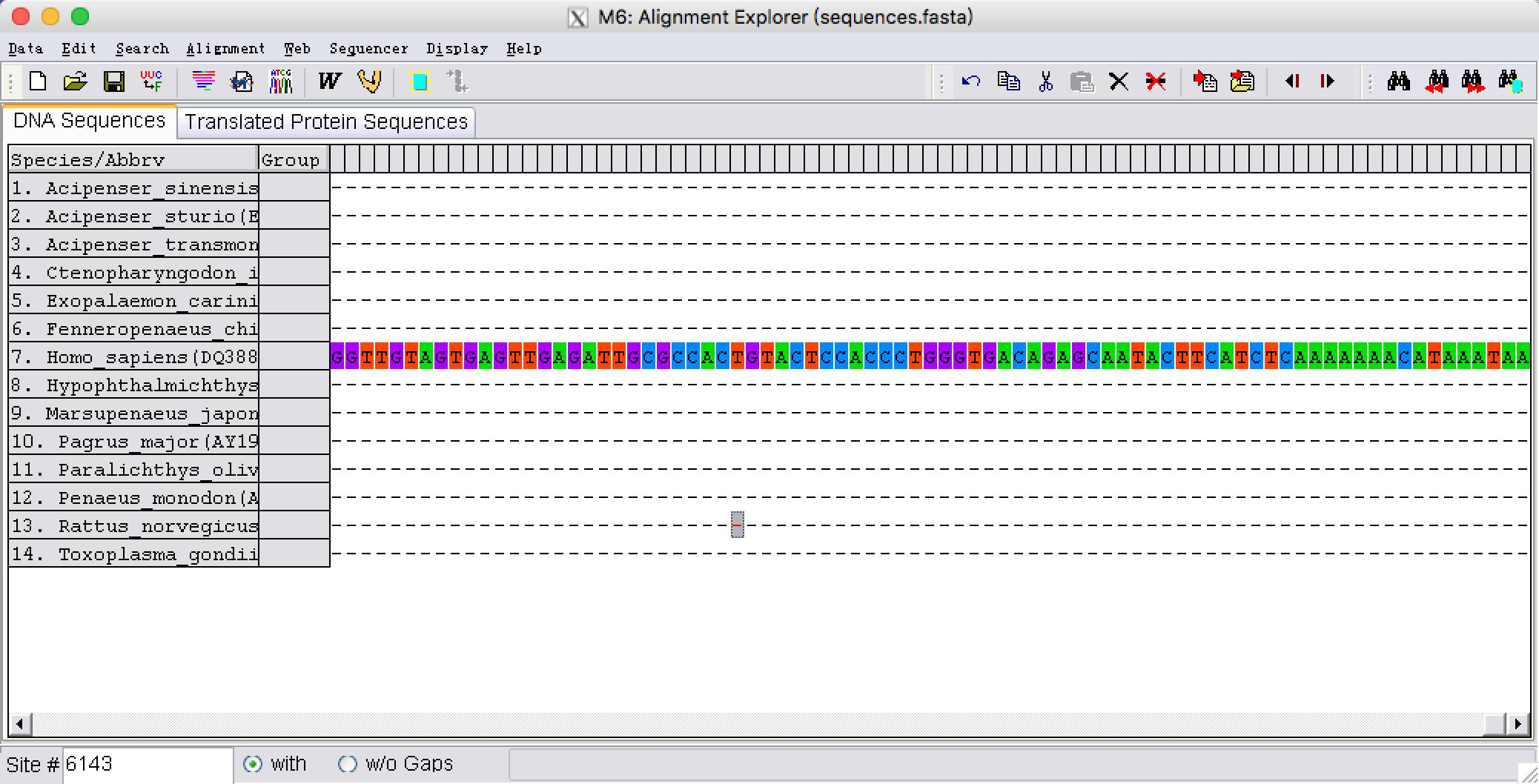
两端对齐之前
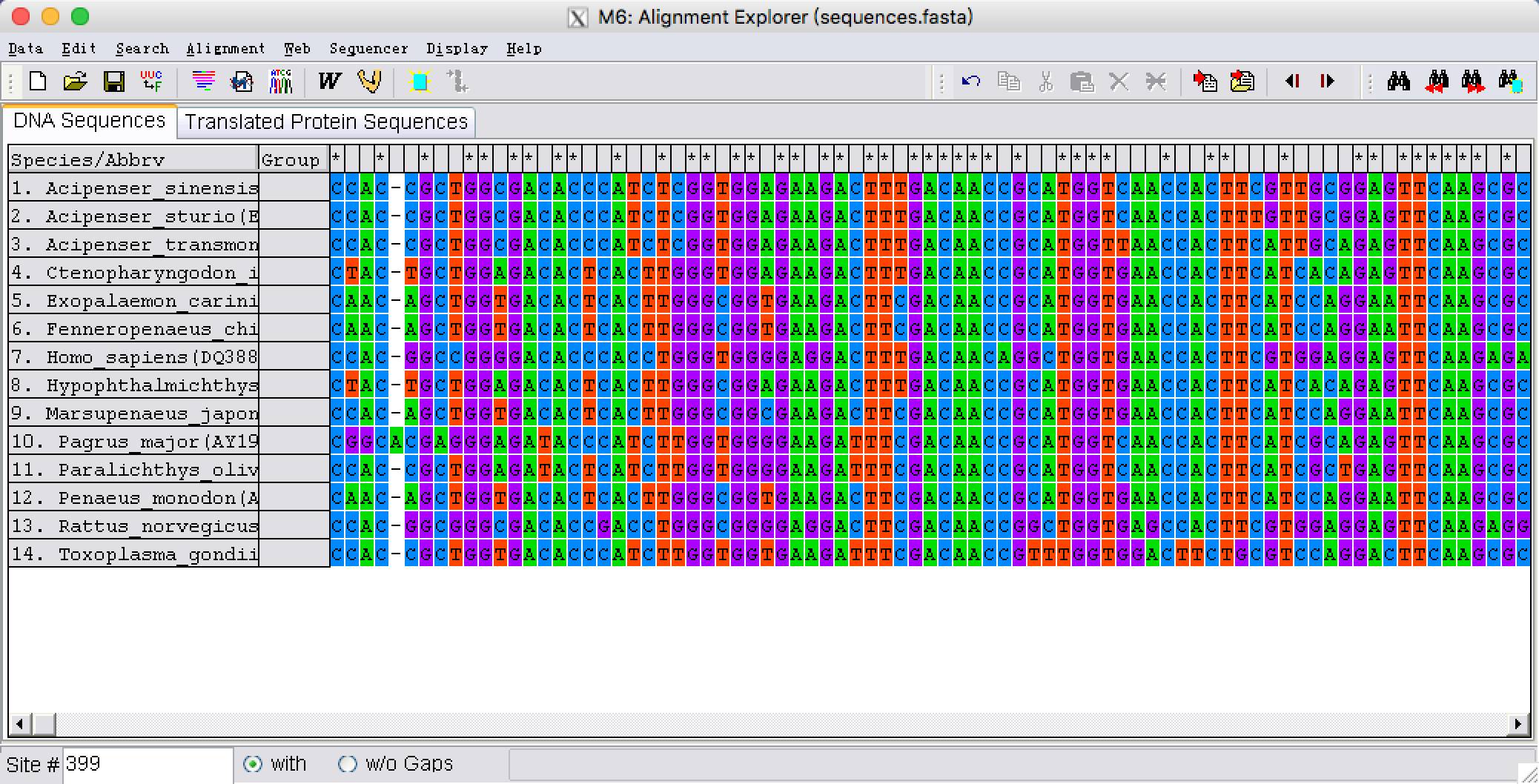
两端对齐之后
6. 然后点Data-Export Alignment-MEGA format,选择一个地方保存,输入Title,关闭多序列比对窗口,点击NO。
7. 点击File,打开刚才保存的.Meg文件

8. 点击Phylogeny,有5个构建进化树的方法,一般选择MaximumLikelihood(最大似然法)Neighbor-Joining(邻接法)和Minimum-Evolution(最小进化法)方法,UPGMA和Maximum Parsimony不常用,点YES,运用当前数据。
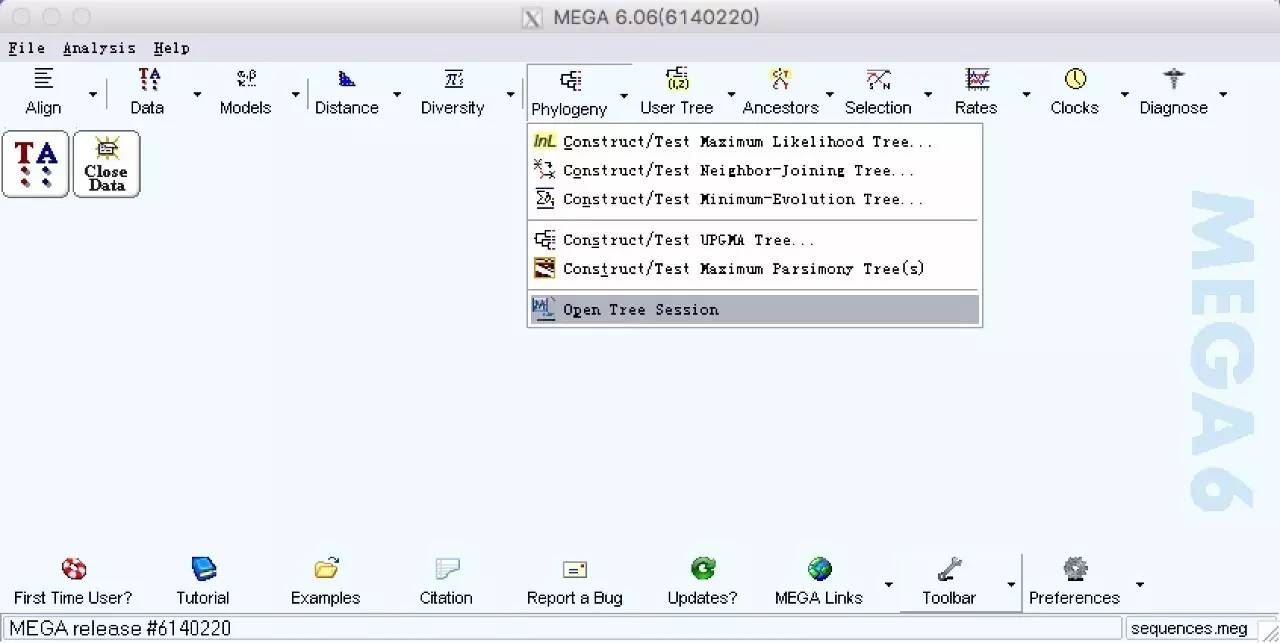
9.在Test of Phylogeny 选择Bootstrapmethod;No.of Bootstrap replications 输入1000;Mode/Method 核酸选择 Maximum Composite Likehood,氨基酸序列选择Poisson model;Rates among sites 选择Uniform rates;Gaps/Missing Data Treatment 选择 Complete deletion. 点击Compute.(一般来说,如果选择同一基因序列长度较一致,物种间情缘关系较近,每种模型构建的进化树差别不会很大。)
10.选择在orginal tree 中显示进化树,数字代表bootstrap值,一般bootstrap值>70以上的树具有可信度。工具栏上的不同按钮可以对树进行修饰,如

可以对不同聚类分支命名;

可以交换聚类的位置;

选择树根,一般来说选择进化地位比较远的物种作为树根;

选择树的不同表现形式。
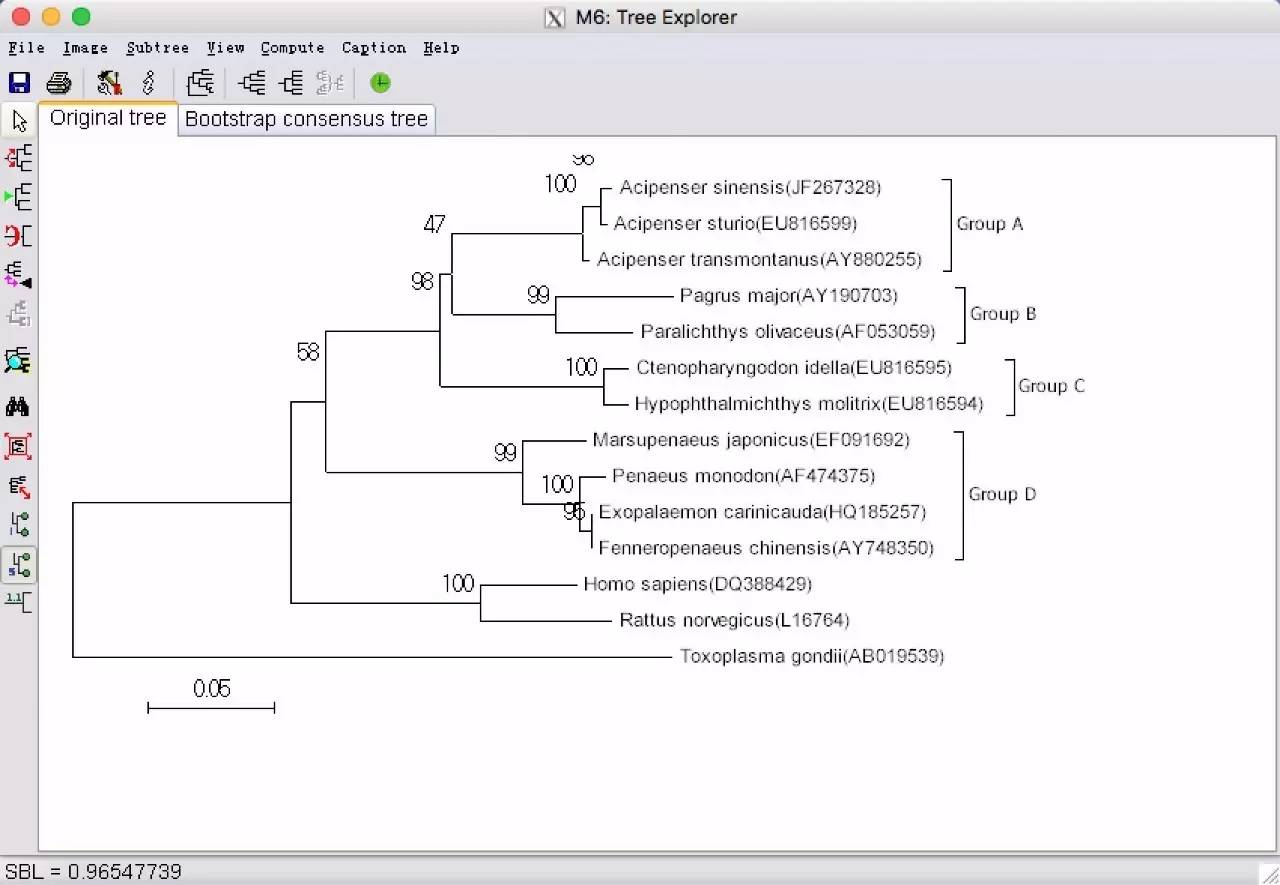
妥妥的tree来了,会了么?最后在Image下的Save as 保存,有3种不同的图片格式(.EMF、.PNG和.PDF)。如果还需要对聚类图用其它软件进行修饰,建议保存为.EMF格式。
来源:解螺旋
2016-05-19